
The Role of Peptides in Alzheimer’s Disease
Alzheimer’s disease (AD) represents one of medicine’s most challenging neurodegenerative disorders, progressively destroying memory and cognitive function in millions worldwide. At the molecular level, two key peptides drive its devastating effects: amyloid-β (Aβ) and tau. These peptides undergo abnormal changes in the brain, with Aβ forming plaques outside neurons and tau creating tangles inside them. According to the “amyloid cascade hypothesis,” the deposition of Aβ peptides initiates a series of events leading to tau hyperphosphorylation, synaptic dysfunction, and neuronal death. Alongside Aβ, tau peptides—normally associated with microtubule stabilization—undergo pathological modifications that result in the formation of neurofibrillary tangles, further impairing neuronal function. The misprocessing of these specific peptides represents a consistent pathological hallmark of the disease, even as researchers recognize its multifactorial etiology.
This review examines the critical roles these peptides play in AD, from their normal biological functions to their transformation into toxic aggregates. We analyze recent breakthroughs in understanding how these peptides interact, trigger neurodegeneration, and ultimately lead to cognitive decline. Understanding these peptide dynamics is crucial for two primary reasons: it provides insight into the molecular mechanisms of neurodegeneration and opens potential therapeutic windows through targeted interventions. Our discussion extends to promising therapeutic approaches targeting these peptides, including novel strategies for early detection and intervention that could reshape our approach to treating this devastating disease. This article synthesizes recent findings on peptide dynamics in AD and evaluates emerging strategies to halt or reverse disease progression.
Peptide Biology in Alzheimer’s Disease
Amyloid-β Peptides : Generation and Processing
Amyloid-β peptides are derived from the amyloid precursor protein (APP) through sequential proteolytic cleavages. APP can be processed via two major pathways:
- Non-Amyloidogenic Pathway: Involves cleavage by α-secretase, precluding the formation of Aβ peptides.
- Amyloidogenic Pathway: Involves sequential cleavage by β-secretase (BACE1) and γ-secretase, yielding peptides of varying lengths, predominantly Aβ40 and Aβ42. Notably, Aβ42 is more prone to aggregation due to its hydrophobic nature and is considered the principal isoform contributing to plaque formation.
Aggregation and Neurotoxicity
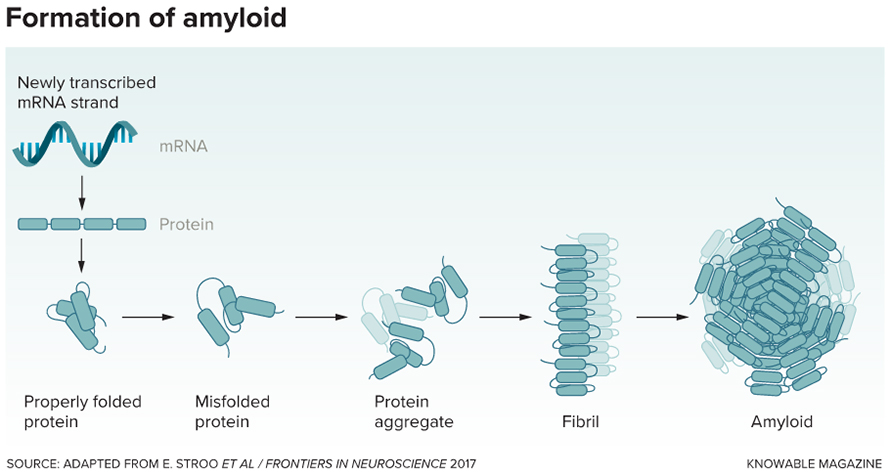
Under normal conditions, amyloid-β (Aβ) peptides exist primarily as soluble, single molecules in the brain. However, in Alzheimer’s disease, these peptides undergo a dramatic transformation, that causes them to misfold into complex structures: first forming small clusters (oligomers), then larger assemblies (protofibrils), and finally insoluble fibrils that accumulate as the characteristic amyloid plaques. Recent research has revealed that the smaller and soluble oligomers, rather than the visible plaques, appear to be the most dangerous forms as they act as molecular toxins that disrupt communication between neurons. These toxic oligomers set off a cascade of destructive events in the brain that trigger oxidative stress and the breakdown of synapses, thus driving the persistent progression of Alzheimer’s disease.
Tau Peptides and Neurofibrillary Tangles
Normal Function of Tau
The tau protein plays a vital role in maintaining the brain’s cellular architecture by binding to and stabilizing microtubules. These microtubules act as essential scaffolding for nerve cells and create tracks for delivering nutrients and cellular components throughout their long extensions. Under healthy conditions, tau’s activity is precisely controlled through a process called phosphorylation, which acts like a molecular switch, allowing tau to dynamically attach and detach from microtubules as needed for normal cell function.
Pathological Modifications
In AD, tau becomes abnormally hyperphosphorylated, which diminishes its affinity for microtubules and promotes its aggregation into paired helical filaments. These filaments eventually form neurofibrillary tangles, disrupting intracellular transport and contributing to neuronal dysfunction and death. The interplay between Aβ and tau pathologies is an area of active research, with several studies suggesting that Aβ aggregation may precipitate or exacerbate tau pathology.
Other Peptides in Alzheimer’s Disease
Neuropeptides and Inflammatory Mediators
Beyond Aβ and tau, other peptides and small proteins play modulatory roles in AD. Neuropeptides such as somatostatin and neuropeptide Y have been implicated in modulating synaptic activity and may influence peptide clearance mechanisms. Additionally, peptides involved in inflammatory cascades, such as cytokines and chemokines, can exacerbate the neurodegenerative process by activating microglia and promoting chronic neuroinflammation.
The APP Intracellular Domain (AICD)
Following APP cleavage, the APP intracellular domain (AICD) is released and translocates to the nucleus, where it may participate in transcriptional regulation. Although its precise role in AD pathology remains under investigation, alterations in AICD-mediated signaling pathways could contribute to disease progression by affecting gene expression relevant to neuronal survival and inflammation.
Molecular Mechanisms Underlying Peptide Pathology
Protein Misfolding and Aggregation
A central tenet of AD pathology is the misfolding of peptides, leading to a cascade of aggregation events. Misfolded Aβ peptides exhibit a propensity to form beta-sheet-rich structures that aggregate into toxic oligomers. Similarly, tau hyperphosphorylation destabilizes its native conformation, promoting self-aggregation. These aggregation processes are influenced by factors such as genetic mutations and alterations in metal ion homeostasis.
Cellular Stress and Synaptic Dysfunction
Aggregated peptides can induce endoplasmic reticulum (ER) stress and activate apoptotic pathways. Moreover, soluble Aβ oligomers have been shown to interfere with synaptic plasticity, which is essential for learning and memory. Disruption of synaptic function is compounded by tau pathology, which impairs axonal transport and compromises neuronal connectivity.
Neuroinflammation
Chronic activation of microglia and astrocytes in response to aggregated peptides leads to sustained neuroinflammation. This inflammatory milieu not only exacerbates peptide aggregation but also contributes to neuronal injury. The bidirectional relationship between peptide pathology and neuroinflammation creates a vicious cycle that accelerates disease progression.
Therapeutic Strategies Targeting Peptides
Modulation of APP Processing
Targeting the enzymatic processes that generate Aβ peptides represents a promising therapeutic avenue. Inhibitors of β-secretase (BACE1) and modulators of γ-secretase have been investigated extensively. The goal is to shift APP processing towards the non-amyloidogenic pathway, thereby reducing the production of pathogenic Aβ peptides.
Immunotherapy Approaches
Immunotherapeutic strategies aim to enhance the clearance of aggregated peptides. Both active (vaccination) and passive (monoclonal antibodies) immunotherapies have been developed to target Aβ aggregates. Clinical trials have shown mixed results, and ongoing research seeks to optimize these approaches to maximize efficacy while minimizing adverse effects.
Tau-Targeted Therapies
Given the critical role of tau pathology in AD, efforts are underway to develop compounds that inhibit tau hyperphosphorylation. Antisense oligonucleotides and immunotherapies targeting tau are under active investigation. These strategies offer the potential to stabilize microtubule dynamics and restore neuronal function.
Combination Therapies and Future Directions
Considering the interplay between Aβ and tau pathologies, combination therapies targeting both peptides simultaneously may offer enhanced therapeutic benefits. Furthermore, emerging research on peptide biomarkers holds promise for early diagnosis, enabling timely intervention before irreversible neurodegeneration occurs. Advances in personalized medicine and neuroimaging techniques are expected to further refine these therapeutic approaches.
Future research should focus on elucidating the precise molecular interactions between peptides and neuronal components, to result in targeted interventions and explore the potential of combination therapies. Additionally, integrating multi-omics approaches could provide deeper insights into the regulatory networks governing peptide dynamics in AD.
Conclusion
Peptides play a pivotal role in the pathogenesis of Alzheimer’s disease. The generation, misfolding, and aggregation of Aβ and tau peptides underpin many of the neurodegenerative processes observed in AD, from synaptic dysfunction to chronic neuroinflammation. Although significant challenges persist, ongoing research into peptide biology has paved the way for innovative therapeutic strategies that target these pathogenic molecules. Continued investigation into the molecular mechanisms of peptide aggregation and clearance will be crucial for the development of effective interventions aimed at altering the course of Alzheimer’s disease.
References
Hardy, J., & Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science (New York, N.Y.), 297(5580), 353–356. https://doi.org/10.1126/science.1072994
Haass, C., & Selkoe, D. J. (2007). Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nature reviews. Molecular cell biology, 8(2), 101–112. https://doi.org/10.1038/nrm2101
Spillantini, M. G., & Goedert, M. (2013). Tau pathology and neurodegeneration. The Lancet. Neurology, 12(6), 609–622. https://doi.org/10.1016/S1474-4422(13)70090-5
Murphy, M. P., & LeVine, H., 3rd (2010). Alzheimer’s disease and the amyloid-beta peptide. Journal of Alzheimer’s disease : JAD, 19(1), 311–323. https://doi.org/10.3233/JAD-2010-1221.